Bangabandhu Sheikh Mujib Medical University Journal
Volume 16, Issue 4, December 2023
BRIEF ARTICLE
Mutation of NPHS1, NPHS2,
WT1, LAMB2, COL4A5 and other genes in children with idiopathic steroid
resistant nephrotic syndrome![]()
Mst. Shanjida Sharmim1![]() , Golam Muin
Uddin1, Afroza Begum1
, Golam Muin
Uddin1, Afroza Begum1![]() , Habibur
Rahman1, Ranjit Ranjan Roy1
, Habibur
Rahman1, Ranjit Ranjan Roy1![]() , Tahmina
Shirin2
, Tahmina
Shirin2![]() , Mohammad
Rashidul Alam3
, Mohammad
Rashidul Alam3![]() , AKM
Muraduzzaman2
, AKM
Muraduzzaman2![]() , Syed Saimul
Huque1
, Syed Saimul
Huque1![]() , Tahmina
Jesmin1
, Tahmina
Jesmin1![]() , Abdullah Al
Mamun1
, Abdullah Al
Mamun1![]()
1Department of Pediatric Nephrology, Bangabandhu Sheikh Mujib Medical University, Dhaka, Bangladesh
2Institute of Epidemiology, Disease Control and Research, Dhaka, Bangladesh
3National Institute of Preventive and Social Medicine, Dhaka, Bangladesh
Correspondence to: Dr. Mst. Shanjida Sharmim, Email: shanjidasharmim@gmail.com
DOI: https://doi.org/10.3329/bsmmuj.v16i4.66671
Received: 3 Jun 2023; Revised version received: 18 December 2023; Accepted: 20 December 2023
Published online: 27 December 2023
![]()
ABSTRACT
Background: Many children with idiopathic steroid resistant nephrotic syndrome have been reported worldwide due to mutation of NPHS1, NPHS2, WT1 and LAMB2 genes. This study aimed to determine the frequency of mutation of NPHS1, NPHS2, WT1, LAMB2, COL4A5 and other genes and their association with renal histopathological patterns of idiopathic steroid resistant nephrotic syndrome patients.
Methods: This cross-sectional study was conducted on 25 patients with idiopathic steroid resistant nephrotic aged 1-17 years in the Department of Paediatric Nephrology, Bangabandhu Sheikh Mujib Medical University, Bangladesh, from July 2017 to June 2018. Next Generation Sequencing and mutation analysis were performed after DNA extraction from patients' venous blood lymphocytes. Histopathological study of renal tissue was done among 17 patients.
Results: A little more than half (56%) of the patients were male. The mean age at the initial attack of nephrotic syndrome was 94.2 months. They mostly had minimal change disease (41%) and IgA nephropathy (12%). One subject had the NPHS2 gene mutation, histopathologically diffuse mesangial proliferative glomerulonephritis, and clinically stage-4 chronic kidney disease. Another subject had the COL4A5 gene mutation and focal segmental glomerulosclerosis. Both were male and had no familial renal disease, consanguinity, or hematuria.
Conclusion: Children with idiopathic steroid resistant nephrotic syndrome showed NPHS2 and COL4A5 gene mutations. Histopathologically, they showed diffuse mesangial proliferative glomerulonephritis and focal segmental glomerulosclerosis.
Keywords: idiopathic SRNS, genetic mutation, nephrotic syndrome, children, Bangladesh
INTRODUCTION
Nephrotic syndrome is the most common childhood glomerular disease which is a triad of hypo albuminemia, oedema, and hyperlipidemia. The incidence of childhood nephrotic syndrome is 1.2-16.9 per 100,000 children/ year.1 Incidence is highest in those of south Asian ancestry compared to European ancestry. Approximately 10-20% of them have steroid resistant nephrotic syndrome (SRNS).2-4 The patient who has no urinary remission within four weeks of prednisolone therapy 60 mg/m2/day followed by three intravenous pulses of methylprednisolone is termed SRNS.5
Response to steroids varies due to various pathophysiological mechanisms. However, this does not fully explain why initial steroid responsive patients become resistant later. Glomerular structural change, specifically in a patient who has podocyte change, is unlikely to respond to steroids. Most of these structural changes occur in genetic forms of SRNS patients. 3 Renal histopathological classification of SRNS mostly shows focal segmental glomerulosclerosis, minimal change disease and diffuse mesangial sclerosis. SRNS may occur as an isolated kidney disease or as a syndromic form. 6 Many SRNS cases occur due to single-gene mutations, leading to profound podocyte dysfunction. 7, 8
An Indian study in 25 children showed NPSH2 gene mutation in 3 patients, and PLCe1, NPHS1 mutation one in each variety. All presented after their first birthday. 9 Another study was conducted on a large cohort of children (n=1783) where 53.9% of them developed SRNS due to a single gene mutation. 10 Thirty percent of SRNS patients who manifested before 25 years of age, a causative mutation was detected in one of the 30 different SRNS-causing genes. These findings revealed that SRNS and focal segmental glomerulosclerosis are not single disease entities but are part of a spectrum of distinct diseases with genetic etiology. 6, 11 In childhood-onset SRNS, the most common mutations are found in nephrin, podocin, and WT1 encoding genes, and some studies showed LAMB2 gene mutation also. These should be screened to help clinical management and genetic counselling. 3 Treatments according to genetic mutation and renal histopathological pattern can avoid the adverse effects of steroids and other immunosuppressive drugs, decreasing the treatment cost. We, however, do not have these data for Bangladeshi children. This study aimed to determine the frequency of NPHS1, NPHS2, WT1, LAMB2, COL4A5 and other genes mutations and their association with renal histopathological patterns in children with SRNS.
HIGHLIGHTS
1. Eight percent of children of idiopathic steroid resistant nephrotic above one year had a genetic mutation in this study.
2. Most patients had minimal change disease and IgA nephropathy.
3. Patients with genetic mutation had diffuse mesangial proliferative glomerulonephritis and focal segmental glomerulosclerosis on renal histopathology.
METHODS
This cross-sectional study was conducted on 25 idiopathic SRNS patients of both sexes aged 1-18 years in the Department of Pediatric Nephrology, Bangabandhu Sheikh Mujib Medical University (BSMMU), Dhaka, Bangladesh, from July 2017 to June 2018. Patients with steroid-responsive nephrotic syndrome (Complete remission within four weeks of steroid treatment), secondary nephrotic syndrome, and age below one year (Congenital and infantile nephrotic syndrome) were excluded. After taking assent from children below 12 years, assent and parental consent of children above 12 years who met the inclusion criteria were initially enrolled in this study. History was taken regarding age, sex, age at onset of disease, consanguinity, family history of renal disease, and hematuria. Physical examination and relevant laboratory investigations were recorded.
Then, 3 mL of venous blood was collected from all patients in a sterile test tube containing EDTA-Na2 and stored in the Department of Biochemistry, BSMMU laboratory, where samples were stored at -800C until DNA extraction. DNA was extracted from peripheral blood lymphocytes using Invitrogen PureLink Genomic DNA Mini Kit (Carlsbad, USA) in the Virology laboratory of the Institute of Epidemiology, Disease Control and Research. After DNA extraction, quality and quantity were checked by thermoscientific Nanodrop 2000 Spectrophotometer (Wilmington, USA). Then, Next Generation Sequencing was performed by HiSeqTM 4000 Illumina sequencer machine (USA) using nephrotic syndromegene panel (IDT primer, Illinois, USA), aligned to the human reference genome (GRCh37/hg19) using BWA programme, analyzed using Picard and GATK version-3.6. Nephrotic syndrome gene panel included DCK4, ARHGDIA, CD2AP, CFH, COQ2, COQ6, CUBN, DGKE, ITGA3, ITGB4, LAMB2, MEFV, MYO1E, NPHS1, NPHS2, PDSS2, PLCE1, PTPRO, SCARB2, SMARCAL1, TTC21B, ACTN4, ARHGAP24, INF2, LMX1B, PAX2, TRPC6, COL4A5 and WT1 genes. Then, mutation analysis was performed.
A renal biopsy was performed among 17 patients (whose parents agreed having a renal biopsy) for histopathological tests.
Statistical analysis
Quantitative variables were presented as mean and standard deviation. Qualitative data were expressed as numbers and percentages.
RESULTS
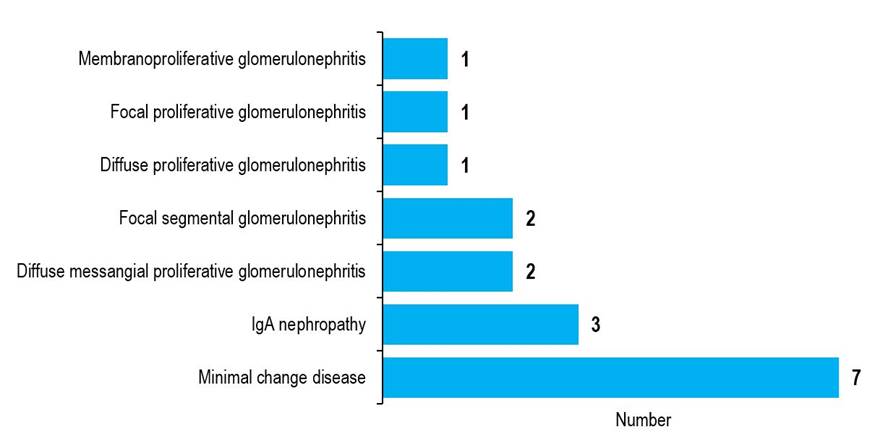
More than half of the participants (56%) were males. The mean age of the study subjects was 106.3 months. The mean age at onset of 1st attack of nephrotic syndrome was 94.2 months. Eighty-eight percent of study subjects were full-term, 12% had a history of consanguinity, 4% had a family history of renal disease, 44% had hematuria, 64% had hypertension, and 56% had pallor (TABLE 1). Their mean serum albumin level was 14.5 gm/ L, serum cholesterol level was 302.9 mg/ dL, 24-hour urinary total protein was 3.9 gm/ day, and serum creatinine level was 1.3 mg/ dL. Histopathologically, 7 (41%) patients had minimal change disease, and 3 (17%) had IgA nephropathy (FIGURE 1).
Only 2 (8%) subjects had a pathogenic mutations (TABLE 2). One had the nonsense type of homozygous mutation of NPHS2 gene in exon 5. The other child had a missense type of hemizygous mutation of the COL4A5 gene in exon 37. The initial attack of nephrotic syndrome occurred at 13 months of age in the NPHS2 gene mutation patient. He was a full-term baby with no family history of renal disease or consanguinity, and renal histopathology showed diffuse mesangial proliferative glomerulonephritis. He had no hematuria but was hypertensive and pale; e-glomerular filtration rate was 22 ml/ min/ 1.73 m2, which corresponded with stage-IV chronic kidney disease.
The initial attack of nephrotic syndrome was at the age of 66 months in a COL4A5 gene mutation patient. He had no family history of renal disease or consanguinity. Renal histopathology showed focal segmental glomerulosclerosis. He had no hematuria or pallor but was hypertensive, and e-glomerular filtration rate was 108 mL/min/1.73m2.
DISCUSSION
In this study, 25 children with idiopathic SRNS had mutations in two genes (NPHS2 and COL4A5). Minimal change disease and IgA nephropathy were the most common subtypes on histological examination.
Thomas et al. showed that the mean age of onset of the disease was 34.8 months.12 However, study participants' mean age and onset age are not directly comparable because of the differences in their enrollment criteria. Many studies, like ours, observed slight male preponderance.13 Most of our study subjects were full-term baby. Congenital nephrotic syndrome usually happens in premature deliveries.14 Lipska et al. showed that 21% had a positive family history with sporadic steroid resistant nephrotic syndrome.14 Studies indicate that the causative recessive mutations are related to consanguinity. 6, 12 and a positive family history. But sporadic mutation in SRNS were present in some studies. 12
Our findings of hematuria,15 hypertension,16 and anemia17 are similar to the studies worldwide. Low serum albumin, high serum cholesterol, significant proteinuria and raised serum creatinine levels12 might have been accentuated by children’s malnutrition, similar to our study. Most of the cases in our series had minimal change disease and IgA nephropathy, identical to other study findings. 12
The present study observed mutations in 8% only. One patient had an NPHS2 gene mutation, and another had a COL4A5 gene mutation. Kari et al. showed that among the 44 children above one year of age with SRNS, five children had pathogenic mutations.18 Among them, 3 cases had NPHS2 gene mutations, and two cases had NPHS1 gene mutations. NPHS2 gene mutation was presented at the age of 12 months, and histopathologically, focal segmental glomerulosclerosis was present.18 Thomas et al. showed that out of 4 NPHS2 gene mutations, one patient had no history of consanguinity or family history of nephrotic syndrome, renal function normal, having membranous glomerulonephritis, a heterozygous missense mutation in exon 4, taking five immunosuppressive drugs. 12 Another study reported 6 mutations (COL4A3, COL4A4, and COL4A5 genes) in 19 patients with genetic mutations.19 Another study showed that a male SRNS patient presented at the age of 16 years with a negative family history or consanguinity, had progressive renal failure, no overt hematuria or hearing loss, FSGS on renal histopathology, and had COL4A5 gene mutation.20
Conclusion
The frequency of identified disease-causing mutation in children older than one year with SRNS was 8%. The identified mutation was present in NPHS2 and COL4A5 genes. Minimal change disease was the major histological pattern. Children with genetic mutations had diffuse mesangial proliferative glomerulonephritis and focal segmental glomerulosclerosis in renal histopathology. NPHS2 gene mutation patient had stage-4 chronic kidney disease.
Acknowledgments
We thank all patients and their parents for their cooperation.
Author Contributions
Conception and design: MSS, GMU, AB, HR, RRR, SSH. Acquisition, analysis and interpretation of data: MSS, GMU, MRA, TJ, AAM, TS, AKMM. Manuscript drafting and revising it critically: MSS, GMU, MRA, SSH, AB. Approval of the final version of the manuscript: GMU, MSS, AB. Guarantor of accuracy and integrity of the work: GMU, MSS, AB.
Funding
The study received partial funding from the Department of Research and Development, BSMMU, and the Akij group of industries.
Conflicts of Interest
The authors declare no conflict of interest.
Ethical Approval
Ethical approval was taken from the Institutional Review Board of Bangabandhu Sheikh Mujib Medical University, vide memo number BSMMU/2017/12019 dated 29 November 2017.
ORCID iDs
Mst. Shanjida Sharmim https://orcid.org/0000-0002-0682-4719
Afroza Begum https://orchid.org/0000-0003-1894-9292
Ranjit Ranjan Roy https://orchid.org/0000-0003-2717-3908
Tahmina Shirin https://orchid.org/0000-0002-5061-3783
Mohammad Rashidul Alam https://orchid.org/0000-0001-8109-5934
AKM Muraduzzaman https://orchid.org/0000-0002-3757-3027
Syed Saimul Huque https://orcid.org/0000-0002-5095-7906
Tahmina Jesmin https://orcid.org/0000-0003-2787-3103
Abdullah Al Mamun https://orcid.org/0000-0001-6308-8159
References
1. Noone DG, Iijima K, Parekh R. Idiopathic nephrotic syndrome in children. The Lancet. 2018 Jul 7;392(10141):61-74. DOI: https://doi.org/10.1016/S0140-6736(18)30536-1.
2. Chanchlani R, Parekh RS. Ethnic Differences in Childhood Nephrotic Syndrome. Front Pediatr. 2016 Apr 19;4:39. DOI: https://doi.org/10.3389/fped.2016.00039.
3. Saleem MA. New developments in steroid-resistant nephrotic syndrome. Pediatric Nephrology. 2013 May;28:699-709. DOI: https://doi.org/10.1007/s00467-012-2239-0.
4. Agrwal S, Mantan M, Dabas A, Batra VV. Childhood steroid-resistant nephrotic syndrome: long-term outcomes from a tertiary care center. Indian Journal of Nephrology. 2022 Jul;32(4):320. DOI: https://doi.org/10.4103%2Fijn.ijn_258_21.
5. Nephrotic syndrome in children: prediction of histopathology from clinical and laboratory characteristics at time of diagnosis. A report of the International Study of Kidney Disease in Children. Kidney Int. 1978 Feb;13(2):159-165. DOI: https://doi.org/10.1038/ki.1978.23.
6. Preston R, Stuart HM, Lennon R. Genetic testing in steroid-resistant nephrotic syndrome: why, who, when and how?.Pediatric nephrology. 2019 Feb;34:195-210. DOI: https://doi.org/10.1007/s00467-017-3838-6.
7. Büscher AK, Kranz B, Büscher R, Hildebrandt F, Dworniczak B, Pennekamp P, Kuwertz-Bröking E, Wingen AM, John U, Kemper M, Monnens L. Immunosuppression and renal outcome in congenital and pediatric steroid-resistant nephrotic syndrome. Clinical Journal of the American Society of Nephrology. 2010 Nov 1;5(11):2075-2084. DOI: https://doi.org/10.2215%2FCJN.01190210.
8. Giglio S, Provenzano A, Mazzinghi B, Becherucci F, Giunti L, Sansavini G, Ravaglia F, Roperto RM, Farsetti S, Benetti E, Rotondi M. Heterogeneous genetic alterations in sporadic nephrotic syndrome associate with resistance to immunosuppression. Journal of the American Society of Nephrology. 2015 Jan 1;26(1):230-236. DOI: https://doi.org/10.1681%2FASN.2013111155.
9. Siji A, Karthik KN, Pardeshi VC, Hari PS, Vasudevan A. Targeted gene panel for genetic testing of south Indian children with steroid resistant nephrotic syndrome. BMC Med Genet. 2018 Nov 20;19(1):200. DOI: https://doi.org/10.1186/s12881-018-0714-6.
10. Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S, Engelmann S, Vega-Warner V, Fang H, Halbritter J, Somers MJ, Tan W, Shril S, Fessi I, Lifton RP, Bockenhauer D, El-Desoky S, Kari JA, Zenker M, Kemper MJ, Mueller D, Fathy HM, Soliman NA; SRNS Study Group; Hildebrandt F. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2015 Jun;26(6):1279-1289. DOI: https://doi.org/10.1681%2FASN.2014050489.
11. Lovric S, Ashraf S, Tan W, Hildebrandt F. Genetic testing in steroid-resistant nephrotic syndrome: when and how?. Nephrology Dialysis Transplantation. 2016 Nov 1;31(11):1802-1813. DOI: https://doi.org/10.1093/ndt/gfv355.
12. Thomas MM, Abdel-Hamid MS, Mahfouz NN, Ghobrial EE. Genetic mutation in Egyptian children with steroid-resistant nephrotic syndrome. Journal of the Formosan Medical Association. 2018 Jan 1;117(1):48-53. DOI: https://doi.org/10.1016/j.jfma.2017.02.012.
13. Pokrajac D, Kamber AH, Karasalihovic Z. Children with steroid-resistant nephrotic syndrome: a single-center experience. Materia Socio-Medica. 2018 Jun;30(2):84-88. DOI: https://doi.org/10.5455%2Fmsm.2018.30.84-88.
14. Lipska BS, Iatropoulos P, Maranta R, Caridi G, Ozaltin F, Anarat A, Balat A, Gellermann J, Trautmann A, Erdogan O, Saeed B, Emre S, Bogdanovic R, Azocar M, Balasz-Chmielewska I, Benetti E, Caliskan S, Mir S, Melk A, Ertan P, Baskin E, Jardim H, Davitaia T, Wasilewska A, Drozdz D, Szczepanska M, Jankauskiene A, Higuita LM, Ardissino G, Ozkaya O, Kuzma-Mroczkowska E, Soylemezoglu O, Ranchin B, Medynska A, Tkaczyk M, Peco-Antic A, Akil I, Jarmolinski T, Firszt-Adamczyk A, Dusek J, Simonetti GD, Gok F, Gheissari A, Emma F, Krmar RT, Fischbach M, Printza N, Simkova E, Mele C, Ghiggeri GM, Schaefer F; PodoNet Consortium. Genetic screening in adolescents with steroid-resistant nephrotic syndrome. Kidney Int. 2013 Jul;84(1):206-213. DOI: https://doi.org/10.1038/ki.2013.93.
15. Gulati S, Sharma AP, Sharma RK, Gupta A. Changing trends of histopathology in childhood nephrotic syndrome. American journal of kidney diseases. 1999 Oct 1;34(4):646-650. DOI: https://doi.org/10.1016/S0272-6386(99)70388-4.
16. Bennett MR, Pordal A, Haffner C, Pleasant L, Ma Q, Devarajan P. Urinary vitamin D-binding protein as a biomarker of steroid-resistant nephrotic syndrome. Biomarker insights. 2016 Jan;11:BMI-S31633. DOI: https://doi.org/10.4137/BMI.S31633.
17. Feinstein S, Becker-Cohen R, Algur N, Raveh D, Shalev H, Shvil Y, Frishberg Y. Erythropoietin deficiency causes anemia in nephrotic children with normal kidney function. American journal of kidney diseases. 2001 Apr 1;37(4):736-742. DOI: https://doi.org/10.1016/S0272-6386(01)80122-0.
18. Kari JA, El-Desoky SM, Gari M, Malik K, Vega-Warner V, Lovric S, Bockenhauer D. Steroid-resistant nephrotic syndrome: impact of genetic testing. Annals of Saudi medicine. 2013 Nov;33(6):533-538. DOI: https://doi.org/10.5144/0256-4947.2013.533.
19. Wang F, Zhang Y, Mao J, Yu Z, Yi Z, Yu L, Sun J, Wei X, Ding F, Zhang H, Xiao H, Yao Y, Tan W, Lovric S, Ding J, Hildebrandt F. Spectrum of mutations in Chinese children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2017 Jul;32(7):1181-1192. DOI: https://doi.org/10.1007/s00467-017-3590-y.
20. Wuttke M, Seidl M, Malinoc A, Prischl FC, Kuehn EW, Walz G, Köttgen A. A COL4A5 mutation with glomerular disease and signs of chronic thrombotic microangiopathy. Clinical Kidney Journal. 2015 Dec 1; 8(6):690-694. DOI: https://doi.org/10.1093/ckj/sfv091.
|
Table 1 Age, sex, clinical and laboratory findings of study subjects (n=25) |
||
|
Characteristics |
Results |
|
|
|
Number |
Percent |
|
Sex, male |
14 |
56 |
|
Pre-term birth |
3 |
12 |
|
Consanguinity |
3 |
12 |
|
Family history of renal disease |
1 |
4 |
|
Hematuria |
11 |
44 |
|
Hypertension |
16 |
64 |
|
Pallor |
14 |
56 |
|
Genetic mutation |
2 |
8 |
|
|
Mean |
Standard deviation |
|
Age (months) |
106.3 |
49.3 |
|
Age of onset of 1st attack (months) |
94.2 |
52.8 |
|
Serum albumin (gm/ L) |
14.5 |
3.2 |
|
Serum cholesterol (mg/ dL) |
302.9 |
97.2 |
|
24-h urinary total protein (gm/ day) |
3.9 |
2.7 |
|
Serum creatinine (mg/ dL) |
1.3 |
1.5 |
|
|
|
Figure 1 Histological subtypes of children with steroid resistant nephrotic syndrome (n=17) |
|
Table 2 Genotype-phenotype and laboratory variables of the mutation-positive study subjects (n=2) |
||
|
Variables |
NPHS2 gene mutation |
COL4A5 gene mutation |
|
Mutation characteristics |
||
|
Transcript |
NPHS2 (-) (ENST00000367615.4) |
COL4A5 (+) (ENST00000328300.6) |
|
Location variant |
Exon 5 |
Exon 37 |
|
Variant |
c.562G>T (p.Glu188Ter) |
c.3319G>C (p.Gly1107Arg) |
|
Zygosity |
Homozygous |
Hemizygous |
|
Inheritance |
Autosomal recessive |
X-linked dominant |
|
Classification |
Pathogenic |
Likely pathogenic |
|
Age at onset of nephrotic syndrome (months) |
13 |
66 |
|
Sex |
Male |
Male |
|
Birth history |
Term |
Term |
|
Family history of renal disease |
Absent |
Absent |
|
Consanguinity |
Absent |
Absent |
|
Renal histopathology |
Diffuse mesangial proliferative glomerulonephritis |
Focal segmental glomerulosclerosis |
|
Hematuria |
Absent |
Absent |
|
Hypertension |
Present |
Present |
|
Pallor |
Present |
Absent |
|
e-glomerular filtration rate (ml/ min/ 1.73 m2) |
22 |
108 |
(c) 2023 The Authors. Published by BSMMU Journal